1.我が国におけるバイオ医薬品関連ガイドライン
我が国におけるバイオ医薬品の承認申請に関して、申請者が、どの程度のレベルでどの程度の量のデータを蓄積すればよいかなどを示すために、我が国では以下の三つのガイドラインが公表されている。
(1) 薬審第243号通知(昭和59年3月30日)
「組換えDNA技術を応用して製造される医薬品の承認申請に必要な添付資料の作成について」 (pdf)
・大腸菌など組換え微生物由来の医薬品
(2) 薬審1第10号通知(昭和63年6月6日)
「細胞培養技術を応用して製造される医薬品の承認申請に必要な添付資料の作成について」 (pdf)
・組換え動物細胞由来の医薬品
・無限増殖系細胞、正常二倍体細胞、ハイブリドーマなどの大量培養により製造される医薬品
・モノクロ-ナル抗体
(3) 都道府県衛生主管部(局)薬務主管課宛事務連絡(平成元年5月)
「薬審1第10号通知に関する質疑応答について」 (pdf)
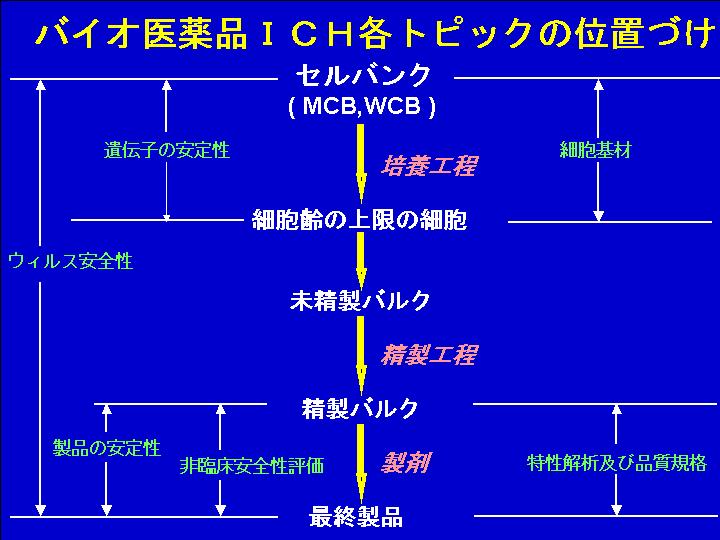
ICHのバイオ医薬品の品質安全性分野では、7つの課題に関するガイドラインが作成されている。すなわち、(1)細胞基材(生産細胞株の適格性と管理)、(2)遺伝子の安定性(遺伝子組換え体の解析と生産培養過程での遺伝子発現構成体の安定性)、(3)ウイルス安全性、(4)製品の安定性、(5)非臨床安全性評価、(6)製品の特性解析および品質規格に関するものである。6つのICHガイドラインは、下図に示すようにバイオ医薬品の製造過程の出発点である細胞基材の問題から培養を経て製品に至り、その品質、安全性の確保に関係する主な課題をカバーしている。さらに最近、申請フォーマットに関する(7)CTD-Qが加わった。 |