Guideline for Bioequivalence Studies for Different Strengths of Oral Solid Dosage Forms
(February 14, 2000)
Index
Section 1: Introduction
Section 2: Terminology
Section 3: Level of formulation change and test required
1. Level of formulation change
2. Test
Section 4: Dissolution test
Section 5: Judgement of equivalence in dissolution
Appendix 1. f2 (similarity factor) and time points
Appendix 2. Normalization of dissolution profiles with lag time
Appendix 3. Level of formulation change and tests
Section 1: Introduction
This guideline describes the principles of procedures of bioequivalence studies for oral solid drug products which are the same in active ingredient, dosage form, therapeutic indication and dosage regimen with a product already approved but differing in strength. The objective of the study is to assure the bioequivalence between products with different strengths when the same doses are administered. The tests required for bioequivalence assessment differ depending on the level of the change in formulation from the approved product.
Section 2: Terminology
Reference product: One lot should be selected from among three marketed lots of products already approved for which therapeutic efficacy and safety were established by clinical trials or bioequivalence was demonstrated by human studies. The reference product should show intermediate dissolution among the three lots under the most discriminative condition, where the difference in dissolution between the fastest and slowest lots is the largest. The dissolution tests (Sec. 4) should be performed using 6 units, by the paddle method at 50 rpm.
Test product: These are products with different strengths from that of reference products. The test product should be manufactured in an production scale or 1/10 production scale or larger. The test product should be the same as the production lots in manufacturing method, quality and bioavailability. In the case of controlled release dosage forms, test products should not significantly differ from the reference product in shape of dosage form, density and release mechanism. The dissolution characteristics of the test product should be similar to those of the reference product as required in the Guideline for Bioequivalence Studies of Generic products (Sec.3.B.1.2) published on December 22, 1997 (http://www.nihs.go.jp/drug/be-guide(e)/be97E.pdf).
Products containing low solubility drugs: When the average dissolution from the reference product does not reach 85% in the testing time specified, that is, 2 hr at pH1.2 and 6 hr at other pHs (pH3.0-7.5), by the paddle method at 50 rpm without surfactants, they are defined as products containing low solubility drugs (see the Guideline for Bioequivalence Studies of Generic products (Sec.3.A.V.3.3).
Section 3: Level of formulation change and test
1. Level of formulation change
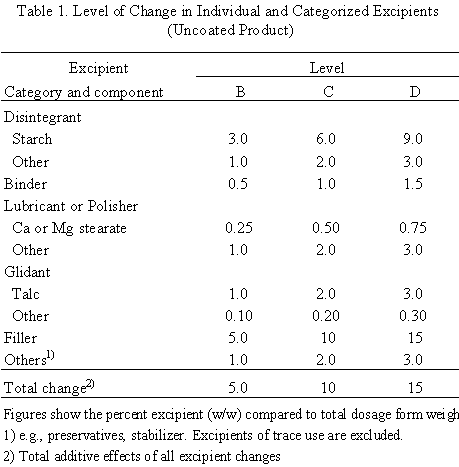
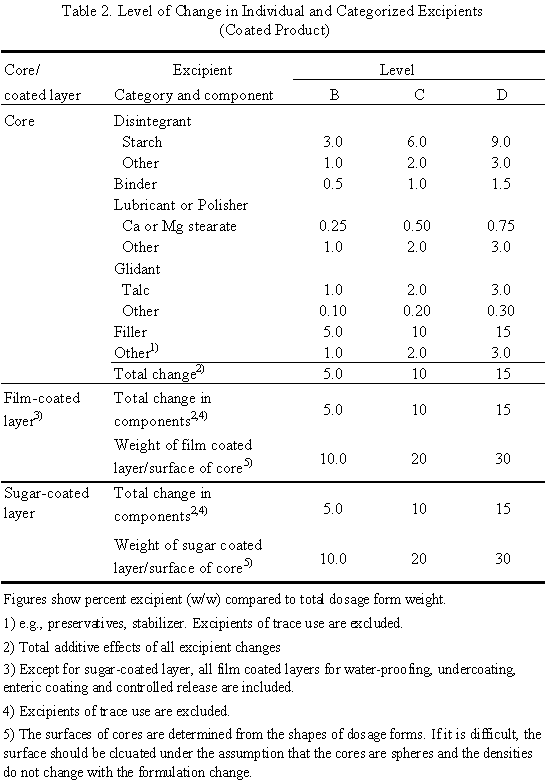
When the ratios of compositions are identical between test and reference products, the formulation change is Level A. This means that test and reference products are the same in ratios of all components including coating agents and, in the case of coated products, the weight of film and/or sugar-coated layers per surface area of the core must be same.
When the ratios are not identical, levels of changes in individual excipients and categorized excipients, shown in Table 1 and Table 2, should be determined. If the change is equal to or less than the ranges of Level B, it is level B. If the change is more than the ranges of level B and equal to or less than the ranges of level C, it is level C. Similarly, the change in excipients in the range between C and D is level D. All changes exceeding the ranges of level D are level E. Any change in excipients whose use is limited to a trace is level A. Among the above changes, the highest level of change is defined as the level of formulation change.
2. Tests
Bioequivalence tests should, in principle, be performed with the same dose, less than the maximum dose shown in the dosage regimen. When the use of different doses is unavoidable, the pharmacokinetic parameters should be normalized for the dose administered which, however, is limited to drugs with linear pharmacokinetics. If more than one dosage unit is used in a dissolution vessel, the drug amount of in the vessel should not exceed the amount of a higher strength product.
Level A
Dissolution tests should be performed using 12 units under the conditions specified in the registration or under the condition shown in Sec. 4 when the dissolution test is not specified. Test and reference products are considered to be bioequivalent when their dissolution is judged to be equivalent according to the criteria in Sec. 5 (1) and (2). If test and reference products are not equivalent in dissolution, bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products.
Level B
Dissolution tests should be performed under the conditions shown in Sec. 4. Test and reference products are considered to be bioequivalent when their dissolution is judged to be equivalent according to the criteria in Sec. 5. If test and reference products are not equivalent in dissolution, bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products.
Level C
Conventional dosage forms and enteric coated products For products containing low solubility drugs, bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products. For other products, dissolution tests should be performed under the conditions shown in Sec. 4. Test and reference products are considered to be bioequivalent when their dissolution is equivalent according to the criteria in Sec. 5, except for narrow therapeutic range drugs listed in Table 3, For narrow therapeutic range drugs, test and reference products are considered to be bioequivalent if their average amounts dissolved at 30 min are equal to or more than 85% under all testing conditions and their dissolution is judged to be equivalent according to the criteria in Sec. 5. If test and reference products do not meet the requirement, bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products.
Controlled release dosage forms For products containing narrow therapeutic range drugs in Table 3, bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products. For other products, dissolution tests should be performed under the conditions shown in Sec. 4. Test and reference products are considered to be bioequivalent when their dissolution is equivalent according to the criteria in Sec. 5, If test and reference products are not equivalent in dissolution, bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products.

Level D
Conventional dosage forms For products containing low solubility drugs and narrow therapeutic range drugs, bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products. For other products, dissolution tests should be performed under the conditions shown in Sec. 4. Test and reference products are considered to be bioequivalent when their average amounts dissolved at 30 min are equal to or more than 85% under all testing conditions and their dissolution is judged to be equivalent according to the criteria in Sec. 5. If test and reference products do not meet the requirement, bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products.
Controlled release dosage form and enteric coated products Bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products.
Level E
Bioequivalence tests should be performed according to the guideline for bioequivalence studies of generic products.
Section 4. Dissolution test
Dissolution tests should be performed according to the conditions shown in Sec.3.A.V and Sec.3.B.I. When polysorbate 80 is added to test fluids for the dissolution tests of products containing low solubility drugs, the concentration should not exceed 0.1%. In the case of enteric coated products, the following test should be added to the dissolution tests specified in the guideline for bioequivalence studies of generic products (Sec.3.A.V);
Paddle method at 50 rpm in 900 ml of pH 6.0 buffer prepared with 0.01mol/L sodium monohydrogenphosphate and 0.005mol/L citric acid.
Section 5. Judgement of equivalence in dissolution
Test and reference products are considered equivalent when they meet both requirements (1) and (2) shown below. The rule is not applicable to conventional dosage forms and enteric coated products, unless the average dissolution from the reference product reaches 85% under any of the testing conditions within the testing time (2 hr at pH1.2 and 6 hr at other pHs) specified in Sec. 3.A.V.2 in the guideline for bioequivalence studies of generic products. When similarity factor, f2 is used, the dissolution data at the time points specified in Appendix 1 should be employed. If dissolution lag is observed for reference products, the equivalence in dissolution can be assessed using the dissolution profile normalized for the lag time. (see Appendix 2)
(1) Average dissolution
(2) Individual dissolution
Test products (n=12) should meet one of the following requirements at the final time points where the average dissolution is compared between test and reference products.
a. When the average dissolution of reference product does not reach 50% within the testing time: There is no sample of test products that shows the deviation of more than 15% in dissolution from the average dissolution of the reference product, and one or no sample that shows the deviation of more than 10%.
b. When the average dissolution of reference product is between 50 and 85 % at the testing time point: There is no sample of test product that shows a deviation of more than 20% in dissolution from the average dissolution of the reference product, and one or no sample that shows a deviation of more than 12%.
c. When the average dissolution of reference product reaches 85 % within the testing time: There is no sample of test product that shows a deviation of more than 25% in dissolution from the average dissolution of the reference product, and one or no sample that shows a deviation of more than 15%.
Appendix 1.
f2 (similarity factor) and time points
(1) Definition of f2
The following equation defines f2, where Ti and Ri show the average percents dissolved from test and reference products at the time point (i), and n is the number of time points.
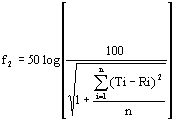
(2) Time point for f2
When there is a lag in dissolution, dissolution data normalized for the lag time should be used for the calculation of f2.
* The testing time is specified in Sec. 3.A.V.2 or Sec. 3.B.I.2 in the guideline for bioequivalence studies of generic products.
Appendix 2.
Normalization of dissolution profiles with lag time
The lag time is conventionally defined as the time when 5% of the drug dissolves. The lag time should be determined for individual dissolution by linear interpolation, followed by normalization of dissolution profiles for the lag time. Then, the average dissolution profiles are determined which can be used for the assessment of equivalence in average dissolution.
Appendix 3.
Level of Formulation Change and Tests
