Guideline for Bioequivalence Studies of
Generic Products
December 22, 1997
Index
Section 1: Introduction
Section 2: Terminology
Section 3: Tests
A. Oral conventional dosage forms and enteric coated products
I. Reference and test products
II. Bioequivalence studies
1. Test methods
1) Design
2) Number of subjects
3) Selection of subjects
4) Drug administration
a. Dose
b. Single vs. multiple dose studies
i. Single dose studies
ii. Multiple dose studies
5) Measurement of drug substances
a. Biological fluids to be sampled
b. Sampling frequency and time
c. Drug substances to be measured
d. Assay
6) Washout period
2. Assessment of bioequivalence
1) Bioequivalent range
2) Parameters to be assessed
3) Logarithmic transformation
4) Statistical analysis
5) Acceptance criteria
III. Pharmacodynamic studies
IV. Clinical studies
V. Dissolution tests
1. Number of units
2. Testing time
3. Testing conditions
1) Products containing acidic drugs
2) Products containing neutral or basic drugs and coated products
3) Products containing low solubility drugs
4) Enteric coated products
4. Acceptance criteria of equivalence of dissolution profiles
VI. Reporting of test results
1. Samples
2. Results
1) Summary
2) Dissolution tests
3) Bioequivalence studies
4) Pharmacodynamic studies
5) Clinical studies
B. Oral controlled release dosage forms
I. Reference and test products
1. Reference products
2. Test products
II. Bioequivalence studies
1. Test methods
2. Assessment of bioequivalence
1) Bioequivalence range, parameters to be assessed, logarithmic transformation and statistical analysis
2) Acceptance criteria
III. Pharmacodynamic and clinical studies
IV. Reporting of test results
C. Nonoral dosage forms
I. Reference and test products
II. Bioequivalence studies
III. Pharmacodynamic and clinical studies
IV. Release tests or physicochemical tests
V. Reporting of test results
D. Dosage forms exempted from equivalence studies
Appendix. List of abbreviations of parameters
Fig. 1a. Bioequivalence test for oral conventional dosage forms and enteric coated products
Fig. 1b. Bioequivalence test for oral controlled release dosage forms
Fig. 1c. Decision of bioequivalence
Fig. 2a. Decision of equivalence in dissolution of oral conventional release dosage forms and
enteric coated products
Fig. 2b. Decision of equivalence in dissolution of oral controlled release dosage forms
Section 1: Introduction
This guideline describes the principles of procedures of bioequivalence studies of generic products. The objective of the study is to assure therapeutic equivalence of generic products to innovator products. In the bioequivalence study, bioavailability should be compared for innovator and generic products. If this is not feasible, pharmacological effects supporting efficacy or therapeutic effectiveness in major indications should be compared (These comparative tests are hereafter called pharmacodynamic studies and clinical studies, respectively). For oral drug products, dissolution tests should be performed, since they provide important information concerning bioequivalence.
Section 2:
Terminology
Terms used in the guideline are defined as follows:
Bioavailability: The rate and extent of absorption of parent drugs or active metabolites from a dosage form into the systemic circulation.
Bioequivalent products: Drug products having the same bioavailabilities.
Therapeutically equivalent products: Drug products having the same therapeutic efficacies.
Innovator products: Products being approved as new drugs by clinical trials or relating drug products.
Generic products: Products whose active ingredients, strengths, dosage forms and regimen are the same as those of innovator's products.
Section 3: Tests
A.
Oral conventional dosage forms and enteric coated products
I.
Reference and test products
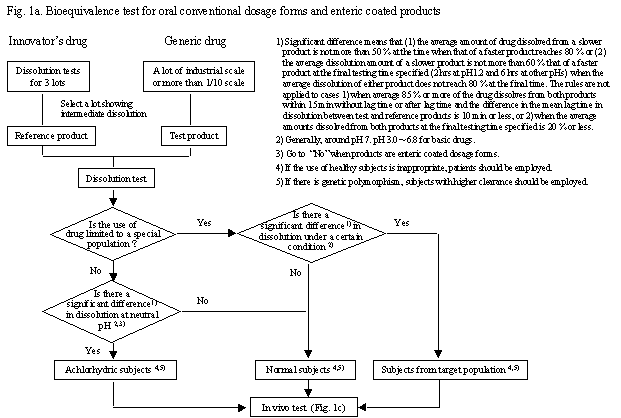
Dissolution tests (Sec. 3.A.V.) should be performed using 6 units or more for three lots of an innovator product by the paddle method at 50 rpm. Among the three lots, the one which shows intermediate dissolution under the most discriminative condition, where the difference in dissolution between the fastest and slowest lots is the largest, should be selected as the reference product. If the dosage form is an aqueous solution or the drug is administered as aqueous solution, any lot of innovator product can be used as the reference product.
As the test product of generic drugs, it is recommended to use a lot of industrial scale. However, a lot of 1/10 or larger of industrial scale can also be used as the test product which should be the same as the production lots in manufacturing method, quality and bioavailability. The drug content or potency of the reference product should be close to the label claim, and the difference in drug content or potency between test and reference products should be less than 5%.
II.
Bioequivalence studies
1.
Test methods
Appropriate study protocol including the required number of subjects and sampling intervals should be determined according to preliminary studies and previously reported data. The rationale of the protocol should be described.
1)
Design
In principle, crossover studies should be employed with random assignment of individual subjects to each group. Parallel designs can be employed for drugs with extremely long half-lives. Replicate crossover studies may be useful for drugs with large intrasubject variability in clearance.
2)
Number of subjects
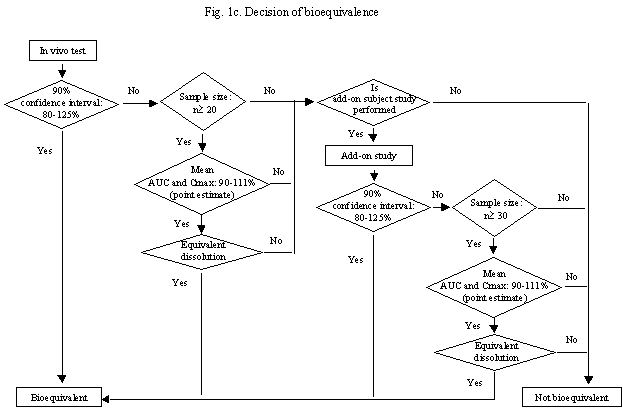
A sufficient number of subjects for assessing bioequivalence should be included. If bioequivalence cannot be demonstrated because of an insufficient number, an add-on subject study can be performed using not less than half the number of subjects in the initial study. A sample size of 20 (n=10/group) for the initial study and pooled size of 30 for initial plus add-on subject study may suffice if test and reference products are equivalent in dissolution and similar in average AUC and Cmax as described under Sec.3 A.II.2.5. Multiple dose studies or studies with stable isotopes may be useful for highly variable drugs that require large sample sizes.
3)
Selection of subjects
In principle, healthy adult volunteers should be employed. When test and reference products showed a significant difference in dissolution* at around pH 6.8 by the dissolution test (Sec.3 A.V) or between pH 3 and 6.8 for products containing basic drugs, subjects with low gastric acidity (achlorhydric subjects) should be employed unless the application of the drug is limited to a special population. This rule is not applied to enteric coated products. If the use of the drug is limited to a special population and test and reference products show a significant difference in dissolution* even under one of conditions of the dissolution test (Sec.3 A.V), the in vivo test should be performed using subjects from the target population.
When it is unfavorable to use healthy subjects because of potent pharmacological action or adverse (side) effects, patients receiving the medication should be employed. If the clearance of drug differs to a large extent among subjects due to genetic polymorphism, subjects with higher clearance should be employed.
Before, during and after studies, subjects' health condition should be monitored with close attention, especially, to adverse (side) effects.
* Significant difference in dissolution means
that 1) the average amount of drug dissolved from a slower product is 50 % or
less at the time when the average amount from a faster product reaches 80 % or
2) the average dissolution amount of a slower product is not more than 60 %
that of a faster product at the final testing time of 6 hr when the average
dissolution of either product does not reach 80 % at 6 hr. This rule is not
applied when average 85 % or more of the drug dissolves from both products
within 15 min without lag time or after lag time where the difference in the
mean lag time in dissolution between test and reference products is less than
10 min. The rule is also not applied when the average dissolution from both
products at the final testing time specified in Sec.V.3 (2 hr in pH1.2 medium
and 6 hr in others) is 20 % or less, because of difficulty of appropriate
comparison of their dissolution.
4)
Drug administration
a. Dose: One dose unit or a clinical usual dose should generally be employed. A higher dose which does not exceed the maximal dose of the dosage regimen may be employed when analytical difficulties exist, such as high detection limit.
b. Single vs. multiple dose studies: In principle, bioequivalence studies should be preformed by single dose studies. Multiple dose studies may be employed for drugs which are repeatedly administered to patients.
i. Single dose studies: Drugs are usually given to subjects with 100-200 ml water (normally 150 ml) after fasting for more than 10 hr. Fasting lasts for at least 4 hr post-dose. If the postprandial dose is specified in the dosage regimen, or if the bioavailability in fasting state is very poor, or high incidence of severe adverse events is anticipated, drugs may be given after food. In the fed study, a low fat diet of 700 kcal or less containing not more than 20 % by energy of the lipid should be employed. The meal should be eaten within 20 min, and drugs are administered according to the dosing regimen or 30 min after the meal, if the dosing time is not indicated in the regimen.
ii. Multiple dose studies: Drugs should, in principle, be administered to subjects under fasting conditions as in the single dose studies when biological fluids are sampled for the assessment of bioavailability. In the time period before fluids are sampled, drugs should repeatedly be given between meals (drugs should be administered more than 2 hr after a meal) at constant intervals.
5) Measurement of drug substances
a. Biological fluids to be sampled: Blood samples should generally be employed. Urine samples may be used if there is a rationale.
b. Sampling schedule: Blood samples should be taken at a frequency sufficient for assessing Cmax, AUC and other parameters. Sampling points should be at least 7, including zero time, 1 point before Cmax, 2 points around Cmax and 3 points during the elimination phase. Sampling should be continued until AUCt is over 80% of AUC\ (normally more than 3 times the elimination half life after tmax). However, when the elimination half life of parent drug or active metabolites is extremely long, blood samples should be collected for at least 72 hr. When urine samples are used, they should be collected in the same manner as blood samples. If F can be determined by deconvolution, such extended sampling may not be required, although the sampling should be continued until drug absorption is complete.
c. Drug substances to be measured: Parent drugs should, in principle, be measured. Major active metabolites may be measured instead of the parent drug, if it is rational. Stereoselective assay is not generally required. However, when it is indicated that there exist stereoisomers with different activities for the main pharmacological effect, and stereoselective absorption or elimination, dependent on the absorption rate is noticeable, the enantiomer with higher activity should be measured.
d. Assay: Analytical
methods should be fully validated regarding specificity, accuracy, precision,
linearity, quantitation limit, and stability of substances in samples and so
forth.
6)
Washout periods
Washout periods in crossover studies between administration of test and reference products should usually be more than 5 times the elimination half life of the parent drug or active metabolites.
2.
Assessment of bioequivalence
1)
Bioequivalence range
The acceptable range of bioequivalence is generally 0.8-1.25 as the ratios of average AUC and Cmax of test product to reference product, when the parameters are logarithmically distributed. The acceptable range is generally -0.2 - +0.2 as the ratio of the relative difference in the mean AUC and Cmax between reference and test products to those of the reference product, when the parameters are normally distributed. For drugs with pharmacologically mild actions, a wider range may be acceptable. The acceptable ranges for other parameters such as tmax should be determined for each drug.
2)
Parameters to be assessed
When blood samples are used, AUCt and Cmax should be subjected to the bioequivalence assessment in single dose studies and AUCt and Cmax in multiple dose studies, Cmax is an observed value and AUC is calculated using the trapezoidal integration method. If F can be estimated by deconvolution, F can be used for AUC.
Parameters such as AUC\, tmax, MRT and kel should be submitted as reference data. For multiple dose studies, Ct also is used as a reference parameter.
When urine samples are used, Aet, Aet, Umax and Ut are employed instead of AUCt, AUCt Cmax and Ct.
3)
Logarithmic transformation
Pharmacokinetic parameters except for tmax should in principle be statistically analyzed after logarithmic transformation.
4) Statistical analysis
The 90% shortest confidence interval or two one-sided t tests with the significance level of 5% should be used. Other reasonable statistical methods also can be used. When add-on subject study is performed and there are no fundamental differences between the two studies in formulation, design, assay and subjects, data from the initial and add-on subject studies can be pooled and statistically analyzed. In the pooled analysis, the study must be added as a source of variation.
5) Acceptance criteria
Products are considered to be bioequivalent, if the 90% confidence interval of difference in the average values of logarithmic AUC and Cmax between test and reference products is within the acceptable range of log(0.8) - log(1.25). However, even though the confidence interval is not in the above range, test products are accepted as bioequivalent, if the following three conditions are satisfied; 1) the total sample size of the initial bioequivalence study is not less than 20 (n=10/group) or pooled sample size of the initial and add-on subject studies is not less than 30, 2) the differences in average values of logarithmic AUC and Cmax between two products are between log(0.9) - log(1.11), and 3) dissolution rates of test and reference products are evaluated to be equivalent under all dissolution testing conditions under Sec.3 A.V. However, the 3rd rule can not be applied to slowly dissolving products from which more than 80% of a drug does not dissolve within the final testing time (2 hr in pH 1.2 medium and 6 hr in others) under any conditions of the dissolution tests described in Sec.3 A.V.
Reference parameters should be subjected to statistical assessment. If a significant difference is detected in the parameters between reference and test products, effects of this difference on therapeutic equivalence should be explained.
III.
Pharmacodynamic studies
These studies are performed to establish the equivalence of products using pharmacological activity in humans as an index. This is applied to pharmaceuticals which do not produce measurable concentrations of the parent drug or active metabolite in blood or urine and those whose bioavailability does not reflect therapeutic effectiveness. In the study, it is desirable to compare the efficacy-time profiles. For antacids or digestive enzymes, suitable in vitro efficacy tests can be employed.
The Acceptance criteria of equivalence in this study should be established by considering the pharmacological activity of each drug.
IV.
Clinical studies
These studies are performed to establish the equivalence of drugs using clinical effectiveness as an index. If bioequivalence studies and pharmacodynamic studies are impossible or inappropriate, this study is applied.
The Acceptance criteria of equivalence in this study should be established by considering the pharmacological characteristics and activity of each drug.
V.
Dissolution tests
Dissolution tests should be performed, using a suitably validated dissolution system and assay.
1. Number of units: 12 units or more under each testing condition.
2. Testing time: 2 hr in pH 1.2 medium and 6 hr in other test fluids. The test can be stopped at the time when the average dissolution of reference product reaches 85 %.
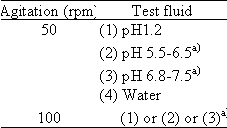
3. Testing conditions: The test should be carried out under the following conditions.
Apparatus: JP paddle apparatus.
Volume of test solution: Usually 900 ml.
Temperature: 37°±0.5.
Test solutions: The 1st and 2nd fluids for the disintegration test (JP13) are used as pH 1.2 and 6.8 test solutions, respectively. Diluted McIlvaine buffers (0.05M disodium hydrogenphosphate/0.025M citric acid).are used for other pH solutions. Other suitable test fluids can be employed when the average dissolution of reference product does not reach 85 % at 6 hr in the McIlvaine buffers.
1) Products containing acidic drugs
a) The test solution should be selected which provides the
slowest dissolution from the reference product and gives average 85%
dissolution or more within the testing time specified, 2 hr at pH1.2 and 6 hr
at other pHs. If the dissolution from reference product does not reach 85 % at
the specified time in any test fluids, the test solution providing the fastest
dissolution should be used.
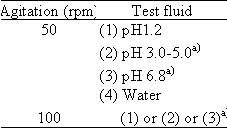
2) Products containing
neutral or basic drugs, and coated products
a) The test solution should be selected which provides the slowest dissolution from the reference product and gives average 85% dissolution or more within the testing time specified, 2 hr at pH1.2 and 6 hr at other pHs. If the dissolution from reference product does not reach 85 % at the specified time in any test fluids, the test solution providing the fastest dissolution should be used.
3) Products containing low
solubility drugs
When the average dissolution from reference product does not reach 85% at the testing time specified (2 hr at pH1.2 and 6 hr at other pHs) at 50 rpm in any of the test fluids, without surfactants, employed in the above dissolution tests 1) and 2), they are defined as products containing low solubility drugs.

a) Among 0.01, 0.1, 0.5 and 1.0 w/w% of polysorbate 80, the lowest surfactant concentration should be chosen that provides average 85% dissolution or more at the testing time specified (2 hr at pH1.2 and 6 hr at other pHs) in at least, one of the test fluids. Dissolution tests in the four fluids should be performed at the same surfactant concentration chosen. If the average dissolution from the reference product does not reach 85% at the specified time in any of test fluids, the surfactant concentration providing the fastest dissolution should be selected.
b) Among the three test solutions, the testing fluid providing the slowest dissolution from the reference product and give average 85% dissolution or more within the testing time specified should be selected. If the average dissolution from the reference product does not reach 85 % at the specified time in any of test fluids, the test solution providing the fastest dissolution should be used.
4) Enteric coated products
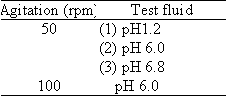
Enteric coated products containing low solubility drugs should be tested by adding polysorbate 80 to the test fluids (2) and (3) according to the dissolution test for products containing low solubility drugs as described above.
4.
Acceptance criteria for equivalence of dissolution profiles
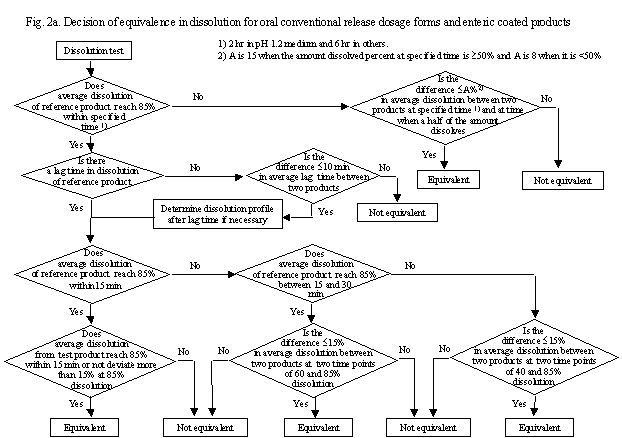
Average dissolution rates of test products should be compared with those of reference products. If the results meet one of the following criteria under all testing conditions, the products are judged to be equivalent. Equivalence in dissolution rate does not necessarily mean bioequivalence.
1) When the average dissolution of the reference product reaches 85% within the testing time specified (2 hr at pH1.2 and 6 hr at other pHs)
a. When there exists no evident lag time* in dissolution of reference products.
i. The average dissolution from reference product reaches 85% within 15 min: the average dissolution from test product also reaches 85% within 15 min or does not deviate by more than 15% from that of the reference product at a time point when the reference product shows approximately 85% average dissolution.
ii. The average dissolution from reference product reaches 85% between 15 and 30 min: the average dissolved amount of the test product does not deviate by more than 15% from that of the reference product at two time points when the average dissolved amount of the reference product is around 60 and 85%.
iii. Others: the average dissolved amount of the test product does not deviate by more than 15% from that of the reference product at two time points when the average dissolved amount of the reference product is around 40 and 85%.
b. When there exists lag time* in dissolution of reference product.
i. The average dissolution from reference product reaches 85% within 15 min after lag time: the difference in average lag time between test and reference products is not more than 10 min. In addition, the average dissolution from test product also reaches 85% within 15 min after lag time or does not deviate by more than 15% from that of the reference product at a time point when the reference product shows approximately 85% average dissolution.
ii. The average dissolution from reference product reaches 85% between 15 and 30 min after lag time: the difference in average lag time between test and reference products is not more than 10 min and the average dissolved amount of the test product does not deviate by more than 15% from that of the reference product at two time points when the average dissolved amount of the reference product is around 60 and 85%.
iii. Others: the average dissolved amount of the test product does not deviate by more than 15% from that of the reference product at two time points when the average dissolved amount of the reference product is around 40 and 85%.
2) When the average dissolution of reference product does not reach 85% within the testing time specified (2 hr at pH1.2 and 6 hr at other pHs)
The average dissolution of test product does not deviate by more than A %** from that of the reference product at the specified time and at the time point when the average dissolution of the reference product is approximately half the specified time.
* lag time: In the case where there exists a delay in dissolution, the lag time is conventionally defined as the time when 5% of the drug dissolves.
** A is 15 when the average dissolution percent is not less than 50% and A is 8 when it is less than 50%.
VI.
Reporting of test results
1. Samples
1) Brand name and lot No. of the reference product. Code No. or name, lot No. and lot size of test product
2) Type of dosage form
3) Name of drug substances
4) Labeled contents or potencies
5) Measured contents or potencies and assay procedures
6) Solubility of drugs at different pHs and in water for dissolution tests
7) Particle size or specific surface area for low solubility drugs and their measurement procedures
8) Types of polymorph and solubility
9) Others (for example, pka and physicochemical
stability)
2.
Results of tests
1) Summary
2) Dissolution tests:
a. List of test conditions (apparatus, stirring speed, types and volumes of test solutions)
b. Assay: method and summary of validation
c. Summary of validation of dissolution tests
d. Results
i. Results of preliminary tests performed to select a reference product.
Tables listing dissolution percent of individual samples under each testing condition, average values and standard deviations of each lot.
Figures comparing average dissolution curves of each lot under each testing condition
ii. Results of preliminary tests performed to select test media.
iii. Comparison of reference and test products
Tables listing dissolved amounts of individual samples under each testing condition, the average values and standard deviations of test and reference products.
Figures comparing average dissolution curves of test and reference products under each testing condition.
3) Bioequivalence studies
Following should be described. Preliminary test items should also be reported.
a. Experimental conditions
i. Subjects:
Age, sex, body weight and other data obtained by laboratory tests are described. Individual gastric acidity should be reported if necessary or otherwise available.
ii. Drug administration
Fasting time, co-administered water volume, and times of drug administration and food ingestion are described. In the case of postprandial administration, menu and content of meal (protein, fat, carbohydrate, calories and others), and times of food ingestion during studies.
iii. Assay: procedure and summary of validation.
b. Results
i. Individual subject data
Tables showing drug levels in biological fluids at each sampling time, Cmax, Ct, AUCt, AUCt, AUC\ kel, tmax and MRT. The correlation coefficient for determining kel should be reported together with time points used.
The ratios of Cmax and AUCt of test product to those of reference product in each individual should be reported.
Figures comparing individual drug level-time profiles of the two products drawn on a linear/linear scale.
ii. Averages and standard deviations
Tables showing averages and standard deviations of raw data of drug levels in biological fluids at each time point, Cmax, Ct, AUCt, AUCt, AUC\ kel, tmax and MRT.
The ratios of average of Cmax and AUCt of test product to those of reference product should be reported.
Figures comparing average drug level-time profiles of the two products drawn on a linear/linear scale.
iii. Statistical analysis and equivalence assessment
Analysis of variance tables for Cmax, Ct, AUCt, AUCt, AUC\ kel, tmax and MRT which are logarithmically transformed when required. The 90% confidence intervals for Cmax and AUCt or alternative statistical results. For other parameters, statistical testing results of the null-hypotheses should be reported where the average values of test and reference products are assumed to be equivalent.
iv. Analysis of pharmacokinetic parameters
If deconvolution is used, the program, algorithm, pharmacokinetic models and fitting information should be listed.
v. Others
Information on dropouts (data, reasons), monitoring records of health of subjects.
4) Pharmacodynamic studies
Reporting of results should follow that of bioequivalence studies.
5) Clinical studies
Reporting of results should follow that of bioequivalence studies.
B.
Oral controlled release dosage forms
I.
Reference and test products
1.
Reference product
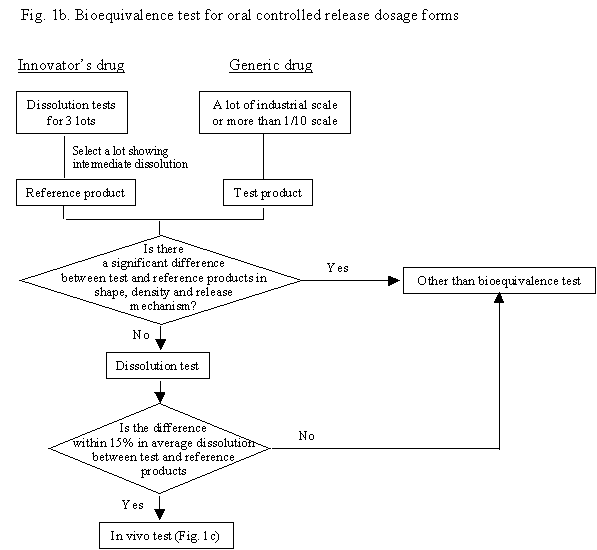
Dissolution tests (Sec.3 A.V) should be performed for three lots of an innovator's product. A lot which shows intermediate dissolution under the condition where the dissolution difference between the fastest and slowest lots is the largest should be selected as a reference product.
2.
Test product
A generic product of controlled release dosage forms should not significantly differ from the reference product in shape, density and release mechanism. The lot size and the drug content or potency follow the same criteria as described in oral conventional dosage forms and enteric coated products (Sec.3 A.I).
The dissolution characteristics of the test product must be similar to those of the reference product under all of the following conditions when dissolution tests are performed according to the dissolution tests for oral conventional dosage forms and enteric coated products (Sec.3 A.V). Either the rotating basket or disintegration testing apparatus can be selected, the reason for which should be stated. The testing times are 2 hr in pH 1.2 medium and 24 hr in other test fluids. The test can be ended at the time when the average dissolution of reference product reaches 85 %.
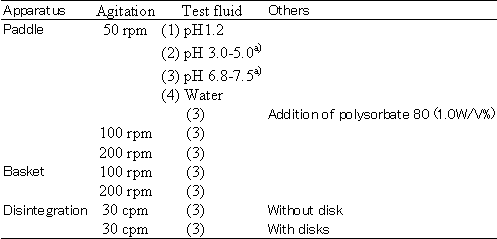
a) The testing solution providing the slowest dissolution in
the pH range which give average 85% dissolution or more within 24 hr should be
selected. If the average dissolution from the reference product does not reach
85 % at 24 hr in any test fluids, the test solution providing the fastest
dissolution should be used.
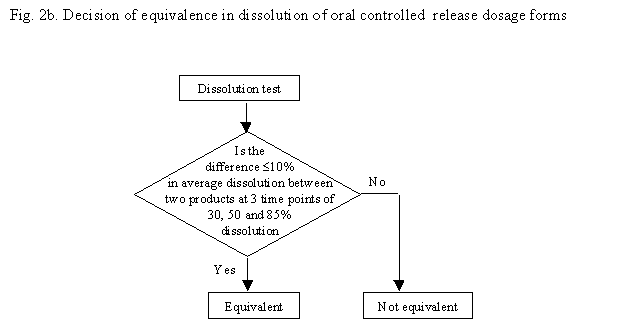
The dissolution profiles are judged to be similar when the average dissolved amount of test product does not deviate by more than 15% from those of reference product under any of the above conditions at three time points when average dissolved amount of reference product is around 30%, 50% and 80%. If the average dissolution of the reference product does not reach 80 % at the testing time specified (2hr in pH 1.2 medium and 24 hr in others), the average dissolved amount at the time specified should be compared.
II.
Bioequivalence studies
1.
Test Method
Bioequivalence studies should be performed by single dose studies in both the fasting and fed states. In the case of postprandial administration, a high fat diet of 900 kcal or more containing 35% lipid content should be used. The meal should be eaten within 20 min, and drugs administered within 10 min thereafter.
When a high incidence of severe adverse events is indicated after dosing in the fasting state, the fasting dose studies can be replaced with postprandial dose studies with the low fat meal employed in the study for oral conventional dosage forms and enteric coated products (Sec. 3. A.II.1.4)). Other testing conditions should follow those of oral conventional dosage forms and enteric coated products (Sec. 3. A.II.1.4)).
2.
Assessment of bioequivalence
1) Bioequivalence range, parameters, data
transformation and statistical analysis
These are the same as those of oral conventional dosage forms and enteric coated products (Sec. 3. A.II.2).
2)
Acceptance criteria
The 90% confidence interval of difference in average value of logarithmic AUC and Cmax between test and reference products is within the acceptable range of log(0.8) -log(1.25).
However, even though the confidence interval is not in the above range, test products are accepted as bioequivalent, if the following three conditions are satisfied; 1) the total sample size of the initial bioequivalence study is not less than 20 (n=10/group) or pooled sample size of the initial and add-on subject studies is not less than 30, 2) the difference in average values of logarithmic AUC and Cmax between two products is between log(0.9) - log(1.11), and 3) the average amount dissolved from test product does not deviate by more than 10% from that of reference product under any of the above test conditions at three time points when the average amount dissolved from reference product is around 30%, 50% and 80%.
The assessment of reference parameters follows that of oral conventional dosage forms and enteric coated products (Sec. 3. A.II.2).
III.
Pharmacodynamic studies and clinical studies
If bioequivalence studies cannot be performed, pharmacodynamic or clinical studies should be carried out to evaluate equivalence according to the studies for oral conventional dosage forms and enteric coated products (Sec. 3. A.III and IV).
IV.
Reporting of test results
The shape, density and release mechanism of the test product should be described which do not differ significantly from those of the innovator product. The description of other results is the same as that for oral conventional dosage forms and enteric coated products (Sec. 3. A.VI)
C.
Non-oral dosage forms
I.
Reference and test products
Suitable release tests or alternative physicochemical tests should be performed for three lots of an innovator's product from which one lot providing intermediate characteristics should be selected as a reference product. The lot size and drug content or potency follow the same criteria as described for oral conventional dosage forms and enteric coated products (Sec. 3. A.I)
II.
Bioequivalence studies
The test should follow the bioequivalence test for oral conventional dosage forms and enteric coated products (Sec. 3. A.II) but the results of release or physicochemical tests are not used for the assessment of bioequivalence.
III.
Pharmacodynamic and clinical studies
The tests proceed in accordance with oral dosage forms. Animal tests will be allowed for topical drugs (skin etc.) with mild pharmacological effects whose equivalence is difficult to demonstrate in humans. In the pharmacodynamic study, it is desirable to compare the efficacy-time profiles. For bactericides for external use, appropriate in vitro efficacy tests can be employed.
IV.
Release tests or physicochemical tests
Release or physicochemical characteristics should be compared between test and reference products by suitable tests which will vary depending on the dosage form.
V.
Reporting of test results
Reporting should be the same as that of oral conventional dosage forms and enteric coated products (Sec. 3. A.VI)
D.
Dosage forms exempted from equivalence studies
Intravenous aqueous solutions.
Appendix.
List
of abbreviations of parameters